Improvements to automated variant interpretation: Part 1
By Kevin Savage onOur Senior Genome Data Scientist for Rare Disease, Kevin Savage, discusses how continually updating gene-disease associations in PanelApp increases the number of diagnoses.
Automated variant prioritisation is a fundamental aspect of rare disease genomics. Back in 2014, when we were introducing whole genome sequencing (WGS) for rare disease diagnostics in the 100,000 Genomes Project, the initial concern was that WGS would identify an unmanageable number of variants for laboratories to review.
We developed an automated variant triaging framework called Tiering to flag a handful of variants that should definitively be considered by the lab for each case. In parallel, we also calculated Exomiser scores and provided systems for the labs to be able to explore the full genomes if necessary. We call these applications Decision Support Systems, somewhat overloading the term.
Tiering prioritises variants most likely to be clinically relevant for individual patients based on known gene-disease association, predicted variant consequence type, population frequency and variant segregation with the phenotype in the family. The diagram below summarises the workflow used to prioritise small variants in Tiering.
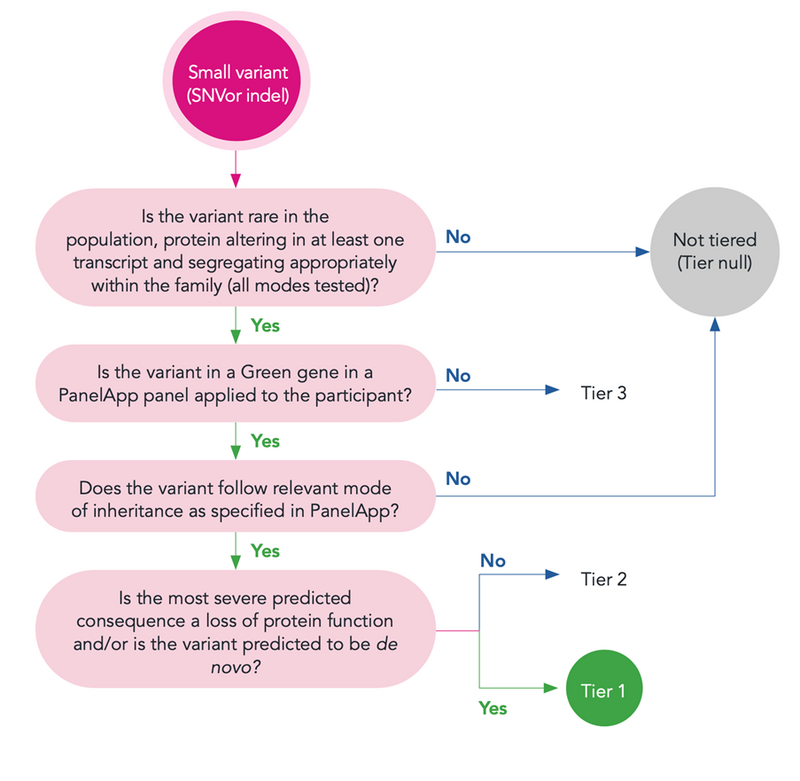
If you are interested in more details on Tiering, see Smedley et al. 2021 NEJM.
Retrospective analysis of non-prioritised variants
Over the course of the 100,000 Genomes Project, we collected information from clinicians about which variants they used in diagnoses. This information was primarily derived from questionnaires completed by clinicians and clinical scientists at the end of the case cycle. From these data, we were able to identify 938 variants that were:
- classified as ‘pathogenic’ and ‘likely pathogenic’ using ACMG criteria,
- considered diagnostic by NHS Genome Medicine Centres,
- not given Tier 1 or 2 priority by Tiering at the time of the first interpretation.
These variants were not identified though Tiering and have been discovered though a variety of other mechanisms, including: Exomiser results (this is also run as part of our pipeline), collaboration with researchers, other external tools such as Decision Support Systems or orthogonal testing.
Using this dataset, we identified the reasons these variants were not prioritised during Tiering, summarised below:

The largest group of variants (50.6%) were excluded because there was not sufficient gene-disease association at the time of analysis, followed by two categories relating to the mode of inheritance (moi) and segregation (fails all seg filters), implying the variant did not meet expected inheritance patterns. The remainder were not prioritised because of the consequence type (i.e., the predicted impact of the variant), high allele frequency, low quality and a very small number not detected at all by WGS. As gene-disease associations was the largest group; we investigated this further.
Impact of improved association data
PanelApp (Rueda Martin et al. 2019, Nature Genetics, 51:1560–1565) is the primary source of gene-disease association used in Tiering. This dataset has improved over time. To investigate the impact of this, we reanalysed the data using updated associations. An additional 17% (166 variants) would be prioritised after reanalysis using current gene-disease association data.
Conclusion
The success of automated variant prioritisation approaches is largely dependent on the accuracy and comprehensiveness of known gene-disease association datasets. Regular updates to this knowledgebase, genotype-lead interpretation and complementary approaches are required and will likely result in continual improvements to diagnostic rates. We also show the value of our ongoing effort to collect information from the GMC outcome questionnaire and how this can feed back into the development of genomic medicine services in the UK.
Using this dataset, we have also been able to show that accommodating complex inheritance patterns with automated variant prioritisation pipelines is particularly challenging. This will be the subject of a future blog piece. Also, watch this blog space for a wider discussion on the approaches adopted at Genomics England to generate and assess the precision of gene panels, which may be highly specific to single disorders or encompass a broad range of clinical presentations.


