One small step for man[ual variant interpretation], one giant leap for genome diagnostics?
By Jamie Ellingford onOur Lead Genome Data Scientist for Rare Disease, Jamie Ellingford, discusses a step towards standardised approaches for variant analysis in non-coding genomic regions.
The 100,000 Genomes Project has accelerated access to the complete genome sequences for families with rare disease, and for the first time for many researchers, enabled access to non-coding regions of the genome for a large clinically characterised cohort.
The non-coding genome, once termed "junk DNA", accounts for ~98% of the 3.4 billion nucleotides that makes up each human genome. As we have learned more about how these regions play an important role in ensuring appropriate regulation and expression of the genetic regions that directly encode proteins, we have also furthered our understanding of genomic variation that disrupts non-coding regions and causes rare human disease (Figure 1).
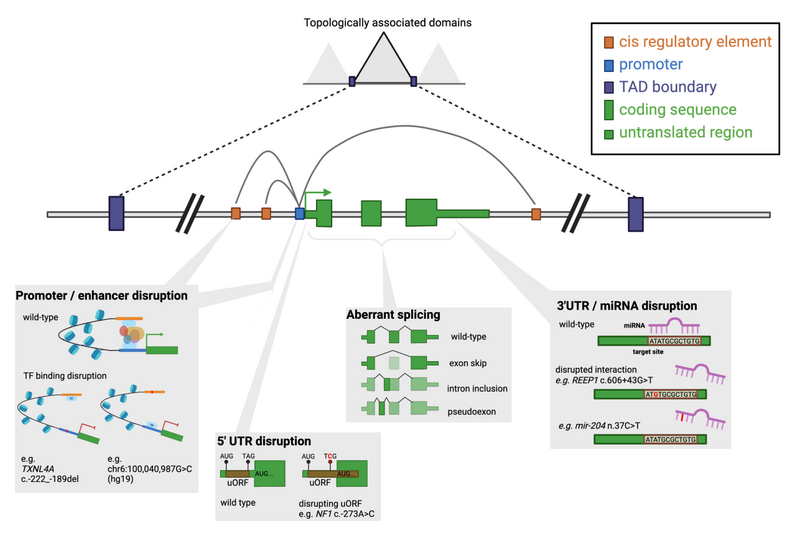
Figure 1. Schematic of key regulatory elements within and around a gene and examples of disruptions that can lead to disease. Extracted from Ellingford et al and created in BioRender.com.
There is now a unique opportunity to discover the diverse mechanisms by which the non-coding genome may be disrupted in disease. However, this is a relatively unexplored area and therefore, until now, there hasn't been a standardised set of rules or recommendations to analyse non-coding region variants in healthcare settings. This blog summarises some of the major messages in a recent manuscript tackling this important issue (standardised recommendations for non-coding region variant analysis, available here) and has been driven through access to the datasets created through Genomics England.
Interrogating non-coding regions of known disease genes will increase the number of individuals with genomic diagnoses
As a community of rare disease researchers, scientists, bioinformaticians and clinicians we have solved many genomic diagnoses for patients and their families. Despite these substantial, and fairly recent, successes in the development of genomic diagnostics, there are some clear learning lessons from existing data: (1) we don’t find genomic diagnoses for everyone with a suspected genomic basis to their disorder; and (2) if we interrogate known disease genes further than what is provided in standard-of-care approaches, we can identify new types of disease-causing genomic variation.
Variation in non-coding regions of the genome is one substantial and diverse area that fits into learning lesson #2 which has received increased attention in recent years. The use of WGS as a frontline diagnostic tool provides a real opportunity to continue to expand these approaches for non-coding region variant analysis to routine healthcare.
The example described below (Figure 2) illustrates the benefit of analysing variants in the non-coding genome for an individual eventually confirmed to have a genetic diagnosis of cystic fibrosis. This was underpinned by the discovery and characterisation of the impact of a variant in a non-coding region of the CFTR gene – CFTR was first discovered as a cause of rare disease in 1989 (Kerem et al), and the first genetic investigations of CFTR for this individual were performed in the 1990s. It was only through interrogation of non-coding regions of CFTR that a conclusive genomic diagnosis could be identified – see [manuscript] for full interpretation of this variant in light of the new guidance.
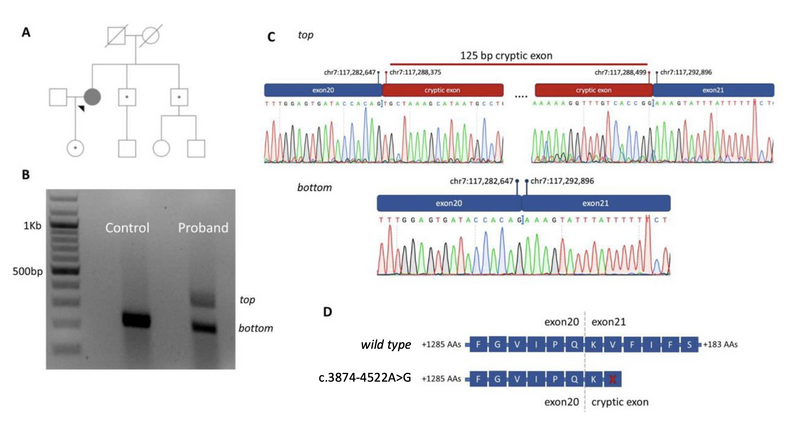
Figure 2. A deep intronic variant (CFTR c.3874-4522A>G) identified in a proband recruited to the 100,000 Genomes Project. (A) Family pedigree showing the proband and unaffected relatives. Carriers of CFTR p.Phe508del are indicated with closed circles within an open symbol. (B) Gel electrophoresis results for the proband and control sample, visualized using a BioRad Universal Hood II. RNA was reverse transcribed after extraction from lymphoblast cell lines and then amplified using primers specific to exons 20 and 21 of the CFTR gene (NM_000492.3). The caption shows two distinct cDNA amplicons in the proband sample separated by ~100 base pairs. (C) Sanger sequencing chromatograms for the CFTR gene generated after cutting out relevant cDNA PCR products from the agarose gel. The larger (top) cDNA PCR product shows a 125 base pair cryptic exon as a result of c.3874-4522A>G. (D) Impact of the cryptic exon on the translated protein. A stop codon is expected to be introduced within the cryptic exon and will result in premature termination of protein synthesis. Amino acids (AAs) are provided with single letter notations, with X indicating a stop codon. Vertical intersects indicate transition of the cDNA to the adjacent exon.
Repurposing existing standards to enable standardised analysis of variants in non-coding regions
Since 2015 the worldwide clinical genomics community has adopted a framework (first set out in Richards et al) to analyse variants in the context of rare disease. This framework enables clinical scientists to assimilate an evidence-base to classify genomic variation on a five-point scale: Benign, Likely Benign, Variant of Uncertain Significance, Likely Pathogenic, Pathogenic.
The non-coding variant recommendations build upon these guidelines to: (1) declare recommendations inappropriate for non-coding region variants (6 rules), (2) leave recommendations exactly as they are (13 rules) or (3) provide specific guidance to repurpose the recommendation to make it appropriate for non-coding regions (9 rules).
For a full description of these changes it is advised that readers go straight to the source of information. The remainder of this blog will give a brief overview of some of the highlights.
Defining appropriate non-coding regions
A pre-requisite for analysing regions of the non-coding genome is an understanding of the non-coding regions that are important for regulation and correct expression of known disease genes. In other words, we need to know about the genomic toolkit required to switch on and control these genes before we can interpret non-coding region variation appropriately.
With the development of highly curated transcript models (e.g. through the MANE project, Morales et al) there is increased understanding of which regions comprise the introns and untranslated regions of known disease genes. However, other non-coding regions such as promoters and cis-regulatory elements are less well defined and not as openly accessible. The new recommendations set out guidance as to how these regions can be characterised along with associated challenges and limitations of defining elements that may be specific to cell-types and/or developmental stages. The cartoon in Figure 1, and descriptions in Table 1, highlight key non-coding regions that the new recommendations attempt to address.
Table 1. Selected classes of non-coding genomic regions that are important for gene regulation and expression.
Type of non-coding region | General definition |
Intron | Non-coding region within the genomic region of a gene that separates distinct exons. Introns are contained within pre-messenger RNA (pre-mRNA) molecules but are excised from the mature messenger RNA (mRNA) via a process called splicing before translation of mRNA to protein. |
5’untranslated region (5’UTR) |
|
3’untranslated region (3’UTR) |
|
MicroRNA (miRNA) |
|
Promoter |
|
Cis-Regulatory Element (CRE) |
|
Topologically associated domain (TAD) |
*The non-coding variant region recommendations do not focus variants that impact TAD boundaries
|
Other evidence codes are more appropriate for non-coding region variants than predicted loss-of-function (PVS1)
Predicted “loss-of-function”, or “LoF” for short, is a prediction that a variant completely knocks out/removes the normal function of the gene that it impacts. Examples include the introduction of premature stop codons (end markers of sequences that are translated from RNA to protein), and large deletion events that impact most of the protein-coding sequence of a gene.
If a variant is expected to cause LoF it can be classified using the original guidelines with the evidence code PVS1. Due to its Very Strong weighting, PVS1 is a highly influential recommendation and should therefore be used with utmost caution and only in situations where it is completely appropriate.
The advice given around non-coding region variants echoes this need for cautious application, and directs analysts towards the use of other rules as more appropriate for consideration (see section below). In particular, the concept that variants in non-coding regions may partially impact gene expression rather than act in a binary (on OR off) manner creates complexity in the use of PVS1 for these purposes.
The increased search space in non-coding regions does not overly impact the occurrence of rare in-trans variants (PM3)
The original recommendation for this piece of evidence enables moderate support for pathogenicity using the evidence code PM3 (as default, although this can be rescaled to other strengths where appropriate) if a rare variant is found in-trans to a well-established pathogenic variant; note this is only applicable for genes associated with recessively inherited disorders.
One significant concern is that the relatively large search space in introns would increase the frequency with which rare non-coding variants are identified in-trans to pathogenic variants (in comparison to in-trans rare variants that impact the protein-coding regions of genes) and make the usage of PM3 inappropriate at the current level of moderate. However, analysis of the 100KGP datasets demonstrated this phenomenon occurs relatively rarely if rare variants are carefully selected (impacting less than 2% of 2016 undiagnosed trio probands) and led to the conclusion, and explicit recommendation, that PM3 can be considered in the same manner for in-trans non-coding regions as has been described for coding region variants.
There is an opportunity (demand) to catalyse the academic and clinical interface with functional investigation!
Given the caution and the limited situations where PVS1 (LoF) should be applied for non-coding region variants, there is a large emphasis on the use of PS3/BS3 – the availability of appropriate functional evidence to support pathogenicity/benignity, respectively. The new guidelines make it intentionally difficult to classify a variant as Pathogenic/Likely pathogenic without the availability of functional evidence for the variant or variants with the same/similar predicted impact. The author group of the manuscript felt that this level of caution was required for the early adoption of these guidelines, and comment that even for some well documented non-coding region variants the level of functional support in the literature is not appropriate for classification as Pathogenic. Whilst this is a limitation of the recommendations, it does enable a unique opportunity for important collaborations across clinical and research multi-disciplinary teams to fully understand variant impact and disease mechanisms, and a requirement to feedback findings from such studies into further refinement of future guidelines. For active research projects in this area using the datasets available in the Genomics England Research Environment browse through the dedicated Research Registry.
Recognising that some very specific, and potentially high-throughput technologies can be used to investigate pathogenicity of non-coding region variants there is specific guidance outlined on the types of investigation and standards that need to be adhered to for use of PS3. Summarising all of these recommendations is beyond of the scope of this blog post, but 2 general words of caution:
- We should not double count evidence – computational tools to predict pathogenicity (PP3) and null impact (PVS1) cannot be used as well as functional evidence (PS3)
- As many functional investigations (PS3/BS3) will investigate levels and specific differences in RNA, understanding the appropriateness of the RNA being surveyed (i.e. is it the correct isoform / are appropriate cis-regulatory elements being used in the study system) and the appropriateness of the datasets that have been generated are very important considerations.
What evidence can be used instead of predicted LoF (PVS1) and in the absence of available functional evidence (PS3/BS3) for the specific variant being analysed?
Three evidence codes have been repurposed for non-coding region variants that may help to fill this void (PM5, PS1 and PM1) - but which code should be used when?
One common trend amongst all three pieces of evidence is that a variant nearby or at the same site must have previously been described as Pathogenic – and as discussed above this will be difficult without appropriate and conclusive functional evidence (PS3). Their application does differ by the specific situations that they try to address, and some specifics are required to illustrate this. The text in italics below summarises the original usage of this rule and is followed by a description of the changes/situations where it is appropriate for usage for variants in non-coding regions:
PM1: mutational hotspots. It is rare that PM1 will be activated for non-coding regions due to the relative importance of specific nucleotides within sequence motifs for many non-coding regions, but situations where PM1 can be activated at a Supporting level of evidence include disruption of specific sub-regions of, e.g., a transcription factor binding site within a cis-regulatory region that has been well documented to be disrupted by variation.
PS1: different nucleotide change results in same consequence (e.g. same amino acid substitution). PS1 has been repurposed in the non-coding region recommendations to be applied at a Supporting level for, e.g., splicing variants predicted to have the same or greater deleterious impact as another splicing variant at the same nucleotide that has been described as pathogenic.
PM5: different nucleotide change results in different but similar impact (e.g. different amino acid substitution at same site which is also deleterious). PM5 can be used for non-coding region variants predicted to have “exactly the same impact, on the same gene, as established pathogenic variants”. E.g. where a different alternative 5’UTR variant is predicted to have exactly the same impact on an untranslated reading frame as a known pathogenic variant in the same 5’UTR.
So there are clear differences in where these rules are appropriate, to summarise:
- PM1_Supporting – can be applied to variants in sub-region hotspots for pathogenic variation within characterised non-coding regions
- PS1_Supporting – limited situations where variants have the same/greater predicted deleteriousness as a known pathogenic, and are at the same nucleotide
- PM5 - limited situations where variants are predicted to have exactly the same impact as a known pathogenic variant, but are not at the same nucleotide
Conclusions
A key reason that these guidelines haven’t already existed is that the data required to analyse the non-coding genome hasn’t been routinely available in diagnostics. Initiatives such as the 100,000 Genomes Project have catalysed the development of a Genomic Medicine Service here in the UK National Healthcare Service, and now the challenge is a reality for clinical scientists. So these guidelines are released at a time – we hope - where the community will really benefit from a standardised way to tackle this multi-faceted challenge.
Whilst the manuscript sets out guidance for the analysis of non-coding regions, there is now an emphasis and requirement for the community to adopt them, test them, and feedback where improvements or refinements should be made. As we discover new pathogenic variants, and as regulatory elements are characterised in greater detail in ever increasing cell types and developmental stages, we expect that these recommendations will also need to evolve and develop.
We look forward to seeing you use the guidelines and to hearing your feedback on their utility.


