Improvements to automated variant interpretation: Part 2
By Kevin Savage onAccounting for more complex inheritance patterns during variant triaging increases number of diagnoses but also increases variant analysis burden
Our Senior Genome Data Scientist for Rare Disease, Kevin Savage, is back with part 2 of this series.
In Part 1 of this series, we discussed the impact of updating gene/disease association data in Genomics England’s variant triaging system, Tiering. In this post we will discuss the impact of incorporating different inheritance patterns for variants within families and outline some of the complications that arise.
In tiering, we can detect if variants are concordant with Mendelian inheritance patterns. For example, we can detect if they are recessive or dominant diseases, or if they are de novo. By comparing this information with the expected patterns of inheritance for specific diseases/genes, we are able to filter out variants that don’t match the expected patterns. This is generally very effective and reduces a large number of variants to a much smaller group that can be reviewed in depth.
However, complexities such as gene and allelic heterogeneity, reduced penetrance, mosaicism, skewed X inactivation, and phenocopies, may cause more complex patterns of inheritance within families and require more sophisticated solutions.
Example – Parental mosaicism
Parental mosaicism (see this Nature article for a good overview) may cause ambiguity between a variant being inherited or de novo. The diagram below shows an example where the mother of the affected child is mosaic and the child has inherited a full 0/1 genotype through the mosaic mother.
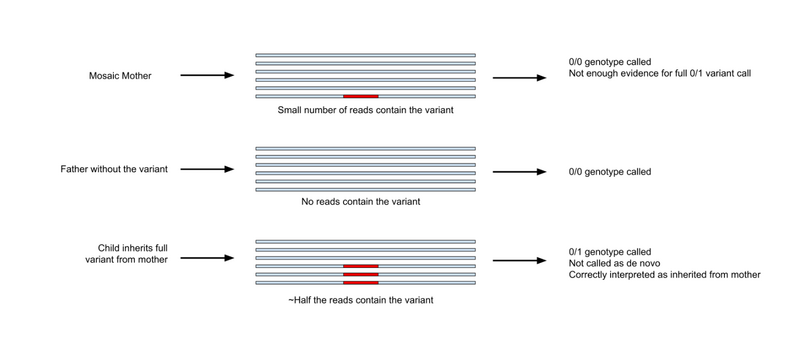
In cases like this, variant callers may give the mother a 0/0 genotype as there is not enough evidence to justify a 0/1 genotype. The child will be given the genotype 0/1 as they have inherited a full allele. However, because of the presence of a small number of reads in the mother, the variant caller may also correctly detect that this variant is not de novo. An additional complexity is that the mother may be affected, not affected or may have a less severe phenotype. These complexities make it difficult to correctly assign a segregation pattern in this family, thereby making it more complex to prioritise variants that fit with the observed inheritance.
Measuring the impact on Tiering
In the last blog, we discussed a dataset of variants we have collected that were:
- classified as ‘pathogenic’ and ‘likely pathogenic’ using ACMG criteria,
- considered diagnostic by NHS Genome Medicine Centres,
- not given Tier 1 or 2 priority by Tiering at the time of the first interpretation.
We used this dataset again to measure the impact of incorporating more complex inheritance patterns into Tiering. To do this, we took advantage of Tiering’s modular design. We were able to use this to change how it treated information about family members. For example, we could simulate a singleton analysis by completely discarding information from samples that were not the proband, thus bypassing expected variant inheritance patterns in the family.
We found that sensitivity can be improved (for this set of individuals) by discarding all information from family members. This recovered 63 out of 191 variants originally deprioritised by segregation or mode of inheritance checks.
We also found a similar improvement in sensitivity when we used reduced penetrance, assuming family members may have the diagnostic variant but not be affected. This recovered 54 out of 191 variants original discarded by segregation or mode of inheritance checks.
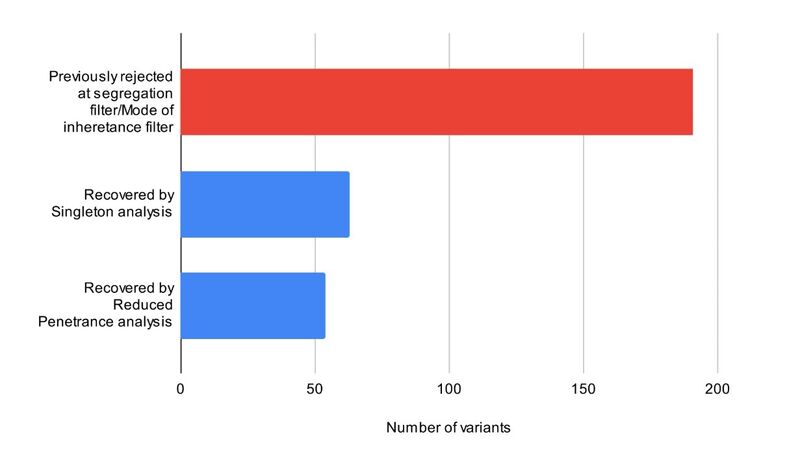
This shows that altering how we use pedigree information during Tiering can result in the recovery of additional diagnostic variants.
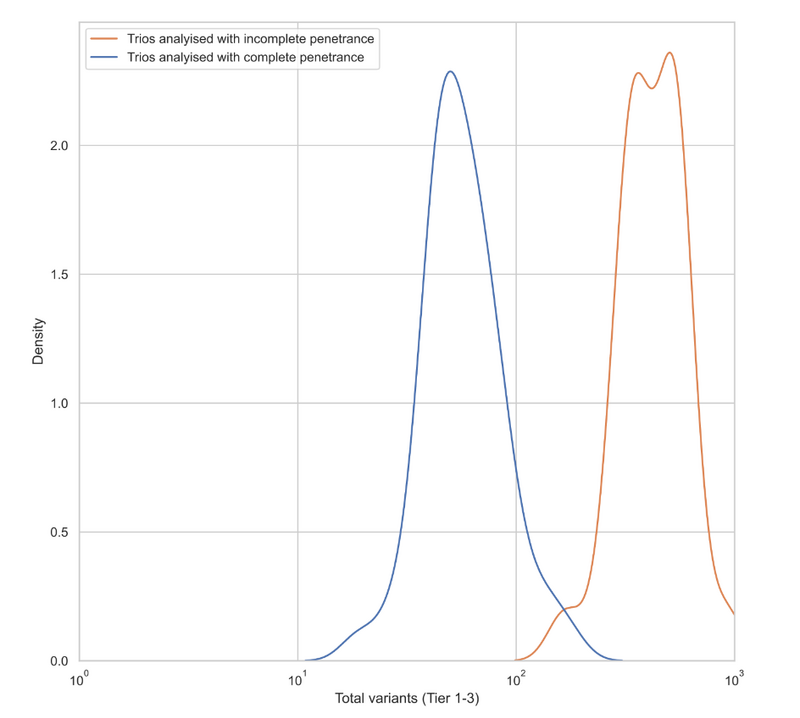
However, whilst this approach has advantages for some individuals, applying it to all individuals referred for genomic testing has a major disadvantage in terms of specificity. The graph below shows the distribution of the number of prioritised variants in a representative sample of trios. Some were originally analysed using complete penetrance and some with incomplete penetrance. As you can see, incomplete penetrance analysis prioritises tens to hundreds more variants per case.
Conclusion
Known gene-disease association data still poses the largest challenge for automated variant prioritisation. However, we also show here that accommodating complex inheritance patterns with automated variant prioritisation pipelines is particularly challenging. We are able to find more diagnostic variants by relaxing the rules applied during variant prioritisation, but at a cost of specificity. Adding more automated algorithms can also be challenging because of the number of genetic hypotheses involved in real cases. For example, it might not be possible to know in advance if reduced penetrance or complete penetrance makes more sense for a particular patient.
We need to continue to engage with the community to ensure that appropriate solutions are put in place to maximise diagnostic yield and limit analysis burdens on clinical scientists – please comment below if you have some thoughts.


