Developing an algorithm to de-identify pathology reports: part 1
By Stanley Ng and Andreia Rogerio onStanley Ng and Andreia Rogerio are applied machine learning engineers at Genomics England. In this role, they investigate and apply new data science and machine learning technologies to improve both internal processes and external user experiences. In part one of this two-part blog series, Stanley and Andreia explain the process of de-identifying personal information and highlight important decisions that were made when designing the pipeline for this.
In October last year, Genomics England made over 40,000 free-text pathology reports from the National Cancer Registration and Analysis Service (NCRAS) available to researchers. These anonymised reports were related to participants in the 100,000 Genomes Project, and include detailed information about tumour types, testing and analysis undertaken in pathology laboratories.
The reports are now uniquely available to researchers in the Genomics England research environment. They exist as a rich dataset alongside other datasets such as diagnosis, treatment and clinical outcome. All of these add to a researcher’s understanding of tumour evolution and progression.
At Genomics England, we put patients and participants at the forefront of everything we do. We therefore made sure that this project would not compromise participant privacy by revealing any of their personally identifiable information (PID).
To ensure that all PID was systematically and efficiently removed from these reports, we developed an algorithmic de-identification pipeline using a combination of pattern-matching and machine learning to fully de-identify each report without any manual intervention.
This pipeline proved as effective as manual review, and successfully de-identified all 40,000+ reports in less than a day, achieving what would have taken a team of reviewers months to process.
In this blog, we’ll explain the design of the de-identification pipeline, how it sits within our quality assurance and data protection systems and highlight some important decisions that we made throughout the project.
How to design an algorithm for de-identification
There are many types of PID. Obvious general ones include people's names and dates of birth, but there are also ones specific to the work we do, such as NHS numbers and disease phenotypes.
We realised early on that each type of PID has its own patterns and levels of risk, so we decided to handle each type separately in our pipeline.
We implemented specific algorithms for each type, such as basic pattern detection logic to detect NHS numbers, (given that each one is simply a 10-digit number). This also allowed us to focus our efforts on information that was more likely to reveal a participant's identity if released, like a participant's name or home address.
Ultimately, separate algorithms for each type of PID allowed us to manage risk more effectively, and to develop and test the components of our pipeline more efficiently.
Our first step was to find out which types of PID were in the pathology reports.
To do this, we read 481 reports ourselves, and labelled all the PID that we found using an open-source labelling tool called Doccano, which came recommended by our collaborators at Hugging Face. We also found that SageMaker Ground Truth from AWS was a useful tool for labelling work, and so we used both over the course of the project.
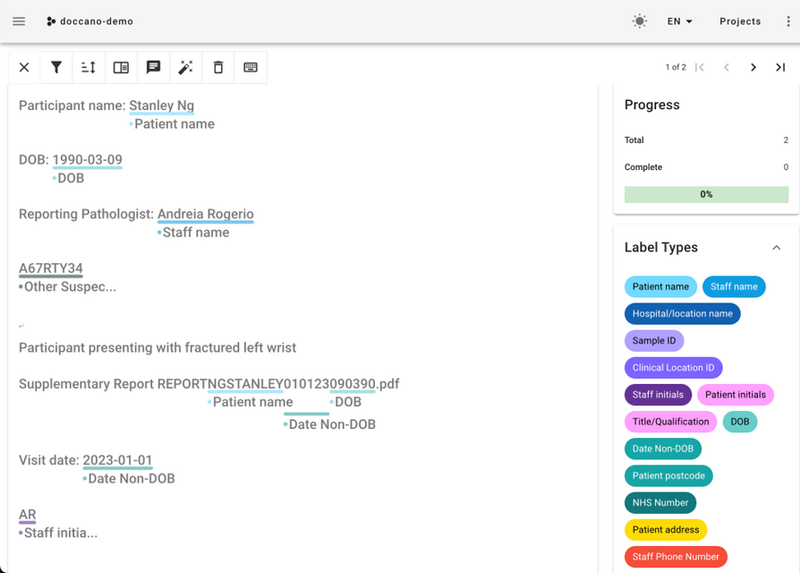
Figure 1: Screenshot from Doccano, one of the labelling tools we used to gain information at the start of the project. Doccano allows sections of text or images to be custom labelled using labels you define (see right). Labels can then be exported for further work, such as exploration or modelling.
What did we need to do, exactly?
With the labels from our 481 reports, we were able to break down the composition of our dataset based on the types of PID that the reports contained. We could also distinguish between PID related to 100,000 Genomes Project participants, which we needed to redact with certainty, from PID related to healthcare staff, hospitals, and other entities.
From these, we identified 3 general strategies that we could use to identify the riskiest types of PID in our dataset.
- Metadata search - In many cases, we have access to relevant personal information related to a participant such as their name and date of birth. In these instances, we can confidently remove that information from any report by simply searching for and redacting all occurrences of the relevant metadata.
- Pattern match - Some types of PID conform to a certain pattern, for example UK postcodes and NHS numbers. It is possible to write pattern-matching logic (using regular expressions, for example) that will capture these types of PID with high specificity.
- Named entity recognition (NER) - Named entity recognition is a natural language processing task in which a model is trained to recognise certain categories of words or phrases from free text. We implemented a pre-trained NER model called FlairNLP in our de-identification pipeline. This was to provide an additional layer of security across all PID types, most crucially for staff-related PID where available metadata is more limited. We will describe our NER approach in more detail in part 2 of this blog series.
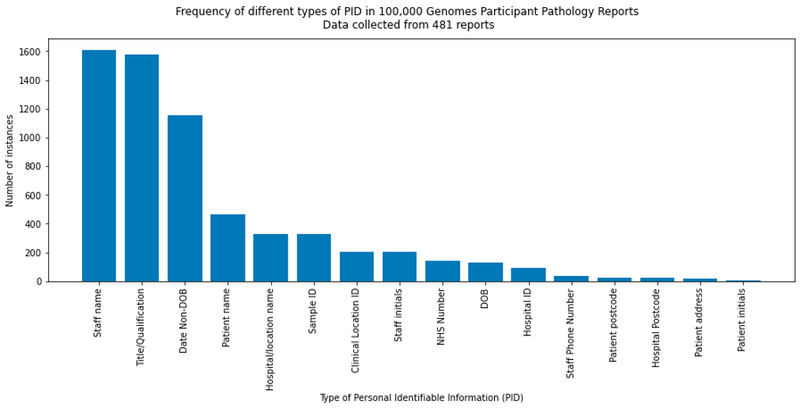
Figure 2: Frequency plot illustrating different types of PID observed in a sub-sample of the pathology reports.
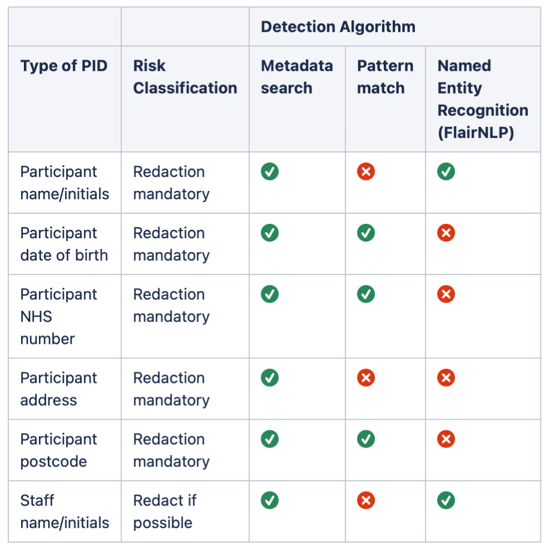
Table 1: Summary of the detection algorithms used to detect each type of PID.
Getting sign-off
Before deploying our solution and pushing data to the research environment, our de-identification pipeline went through several layers of approvals from stakeholders across Genomics England.
In short, everything including the cloud architecture, the data flow and the algorithms themselves were analysed and revised according to feedback from our colleagues in data protection, ethics, platform engineering and other teams. This collaborative effort across Genomics England is crucial to ensure that we provide robust and high-quality products to our users.
Testing the algorithms against human reviewers
Once the pipeline received initial approval, we needed to compare how the pipeline performed against human reviewers on a previously unseen random sample of reports.
Over the course of a week, 4 reviewers read and annotated 600 reports of the ~40,000 in the full dataset. When we compared the pipeline's performance on these 600 reports against the human reviewers, we found that the pipeline had successfully redacted 100% of the mandatory PID (Tables 1 and 2), satisfying the requirement that no personally identifying participant information be left unredacted.
This marked a huge step forward for us. Not only did it mean that we could release the data to the research community, but also that algorithms and machine learning could accelerate our mission to provide a leading environment for genomic medicine research, all while preserving the trust and quality that the community expects from us.
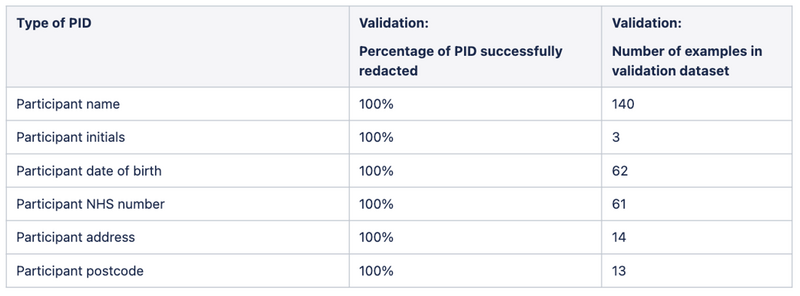
Table 2: Results from our manual validation of the de-identification pipeline, comparing the performance of the pipeline to human reviewers on 600 manually labelled reports.
Next steps
Now that we have released these first 40,000 reports, researchers have already started using the new dataset to enhance their understanding of certain clinical cases in the research environment. This allows them to contextualise other quantitative data in the research environment (e.g. genomic analyses) and imbue their research with additional insight.
We are now exploring expansion of the pipeline scope to de-identify reports for which there is less participant metadata available, for example, by introducing new algorithms and new lookup tables.
We are also investigating whether we can employ NER models to purposefully target and extract clinical indicators from the free text reports, potentially providing researchers with a structured quantitative dataset that was not previously accessible.
We are excited about these opportunities and hope to continue building upon this initial release to deliver more to our research community.
Stay tuned for the second instalment of this series, where we’ll explore details of the pattern-matching and natural language processing algorithms that we used in the pipeline. We’ll explain how and why each of them was chosen.
For now, we’d love to hear your thoughts and questions in the comments section below.
Acknowledgements
Thank you to all the Genomics England colleagues who helped us over the course of this project. In particular, we would like to thank Francisco Azuaje, Prabhu Arumugam, Alona Sosinsky, Georgia Chan, James Micklewright, Marko Cubric, the Multimodal Squad, Geraldine Nash, Geoff Coles, Anna Need, Stuart Ellis, and Roel Bevers for their time, efforts, and insights; without which we would not have been able to complete this work. We would also like to thank Florence Cornish for her help reviewing and improving this blog post. Finally, we would like to thank all the participants of the 100,000 Genomes Project whose data was included here; by continuing to work with us, they help us in our mission to enable and advance research.


