Improving how we report gene fusion productivity
By Nadezda Volkova onNadezda Volkova is a senior cancer analyst at Genomics England with over 5 years of experience in analysing and visualising biological data. In this blog post, she explains how knowing about gene fusions could enhance our understanding of cancer, allowing for improved diagnosis, treatment and, ultimately, cancer outcomes.
What are gene fusions?
Gene fusions are a class of mutation that commonly occur in various cancer types. They typically result from chromosomal rearrangements that cause movement of coding or regulatory regions between genes.
When coding regions are exchanged, the event may produce a Frankenstein-like chimeric protein if the coding sequences are linked correctly. Exchanges in regulatory regions, however, can alter gene expression patterns (Figure 1).
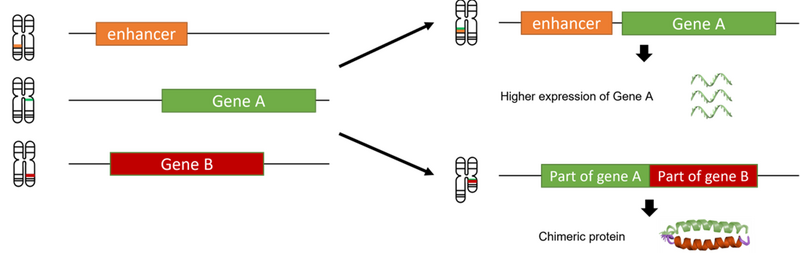
Figure 1. Gene fusions and their consequences
Fusions like this are referred to as ‘productive’, as they have a functional consequence. Some tumours may carry hundreds of variants that bring together different regulatory or coding regions. However, only a small number of these would generate a productive fusion. DNA fragments must come together in the correct order, and the coding sections should be fused in the right place to preserve the triplet reading frame of the genetic code to enable transcription and translation.
This reading frame works for nucleotide sequences in the same way that spaces work to separate words. Take this example, misplacing the spaces can dramatically affect the phrase:
imagine having to read like this
i magineh avingt or eadl iket his
Notice how some words become meaningless or change their meaning completely. In genetic sequence terms, the ends of neighbouring exons should have the same phase – i.e., if one exon finishes after the second letter of a triplet (having end phase 2), the next one should start with the third letter (or have start phase 2), otherwise the protein sequence may change. Such exons can also be called “in-frame”.
When we talk about gene fusions involving two protein-coding genes, both gene partners need to be fused ‘in-frame’ to generate a productive fusion. In these cases, reading through the fusion sequence would still generate a protein consisting of parts of either protein encoded by both fusion partners. If the reading frame of the downstream partner is changed after the fusion point, the fusion is called ‘out-of-frame', and it is unlikely to generate a protein (Figure 2).
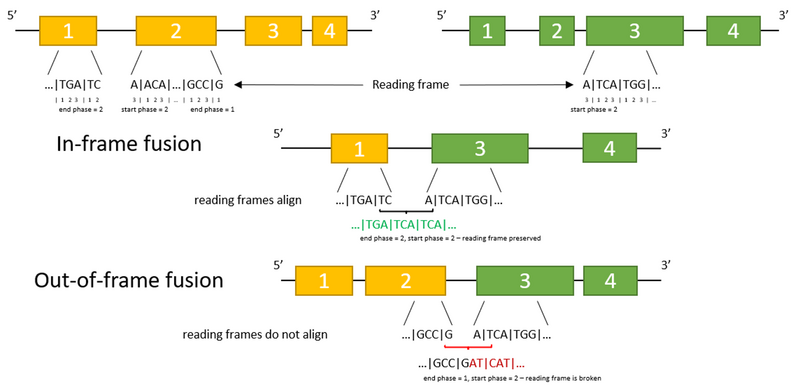
Figure 2. Reading frame and its effect on fusions: in-frame and out-of-frame fusions
What do we want to know about gene fusions?
Gene fusions may be key driver events in tumorigenesis. They provide important information for diagnosis, risk stratification or treatment selection in individuals with cancer.
For example, synovial sarcomas – a rare subtype of sarcoma – is driven by a fusion between SS18 and SSX genes. Discovering this fusion in a tumour and finding out which SSX genes are involved is essential to reach a correct diagnosis.
Some fusions serve as biomarkers that can inform risk or progression, such an example is TMPRSS2-ERG fusion in prostate cancers. Furthermore, certain gene fusions make tumours eligible for targeted therapies, like BCR-ABL1 fusions in leukaemia, or fusions involving ALK, FGFR, or NTRK gene families in solid cancers.
These fusions result in a group of proteins known as tyrosine kinases, which often function as switches for cellular processes such as proliferation, differentiation and programmed cell-death. If mutated or fused with other genes, they can become stuck in the ‘on’ state and cause uncontrolled cell growth. Fortunately, several drugs have been developed that can inhibit tyrosine kinase activity and successfully treat such tumours.
Even if the DNA sequences are perfectly aligned, several downstream events can prevent expression of the hybrid protein. The sequence formed may not be expressed, the RNA expressed from it may be defective and never be translated, or the translated protein may be degraded immediately. There will always be a lot of variants in the tumour, but only a handful will be of consequence.
Testing if gene fusion is present
In clinical practice, there are multiple techniques that can test with varying confidence whether a gene fusion is present in the cell, and whether it produces a corresponding fusion transcript or chimeric protein.
If we know which fusion we are looking for, a technique called reverse-transcription-polymerase chain reaction (RT-PCR) can be applied to see if the fusion mRNA product is present in the cells. Fluorescence in situ hybridization (FISH) could also be used to visually confirm whether genomic regions with known fusion partners occur next to each other.
In addition to these techniques, we can look for hybrid RNA sequences using RNA sequencing (RNAseq) panels, or for the chimeric protein using immunohistochemical staining, but only if we know exactly what to look for. Whole genome RNAseq can also be performed to detect arbitrary fusions, but its results are known to be noisy and often produce false positives.
In short, the most reliable method to know whether a gene fusion is a true productive gene fusion, is to check for the presence of the corresponding chimeric protein.
Computational discovery of productive fusions is difficult but very appealing. Having a way to filter out at least the non-productive events would save a lot of time while interpreting the results of whole genome analysis (WGA), as well as lab resources on alternative testing.
While most efforts have so far been focussed on detecting fusions from RNA sequencing results, several computational tools have been developed over the past years to identify gene fusions from DNA sequencing data. Some of these implement intensive realignment to identify fusion sequences (such as JuLI or Genefuse), while others rely on structural variant calling results (Oncofuse, GRASS, LINX).
All of these methods need to interact with various information sources to query the genes, regions affected, and their phases.
At Genomics England, we have already implemented and validated an extensive variant calling pipeline, so we decided to take the second approach.
Annotating fusion outcomes
Currently, our pipeline reports any variant whose breakpoints affect two different gene coding regions as a potential fusion in whole genome analysis results. Sometimes this leads to hundreds of fusions being reported in a single tumour, all left for the user to interpret.
To make variant review slightly easier, we can apply established knowledge on transcript orientation and the location of different functional elements (UTRs, introns, and exons). We can then annotate DNA variants with an expected outcome should it be transcribed and translated.
Once we have identified a variant and the transcripts it may be affecting, annotating the potential outcome is relatively straightforward. A structural variant can be represented as a set of one or more breakpoints, each breakpoint being a record of connection between two genomic regions. Precise genomic coordinates of those regions are called break-ends (Figure 3).
To assess fusion outcomes, we first look at the orientations of breakpoints. This means we check which side of each break-end the joint sequence continues in (left or right). Such information can be extracted from the VCF file for structural variant calls. Breakpoint orientations for deletions or duplications are always the same, while those for translocations and inversions can vary depending on whether the event was balanced or unbalanced.
Next, we overlap the breakpoint orientations with the transcription directions of the corresponding gene.
Thus, deletions and duplications can only create a productive fusion from partners with the same transcription directions, inversions – from those with opposite transcription directions –and translocations can potentially turn any protein-coding gene pair into a productive fusion depending on the exact breakpoint configuration (Figure 3).
Figure 3. Examples of possible transcription directions of the fusion partners and breakpoint orientations depending on the variant type (BND stands for break-end).
Once we have identified the pairs of transcripts from either side of the breakpoint that could potentially be transcribed in a fusion, we need to check which functional elements are affected by the breakpoints.
Fortunately, this can be done using a query to a database. We annotate each break-end with the functional element of the gene that it falls into: a 5’ UTR intron or exon, coding exons, introns between coding exons, 3’ UTR intron or exon, or – in rare cases – a splice site.
Subsequently, the reading frame outcome is decided using the following logic (Figure 4).
If a fusion involves:
- 3' UTR of the downstream partner, it cannot be transcribed, and the fusion is unproductive
- 5' UTR of the upstream partner, it is denoted as 5' UTR
- 3' UTR of the upstream partner, 5' UTR of the downstream partner, splice sites or any coding exons – we call it ambiguous
- Two introns that are both between coding exons – we can determine whether the exon phases are aligned:
- if yes - it is reported as in-frame
- otherwise - out-of-frame
In addition to in-frame and out-of-frame fusions, we have defined several other outcome classes.
There are many configurations where it is not necessary or feasible to predict the frame. Thus, 5’ UTR fusions are the ones involving 5’ UTR region of the upstream partner – they can result in a change of expression of the downstream partner, but no fusion transcript.
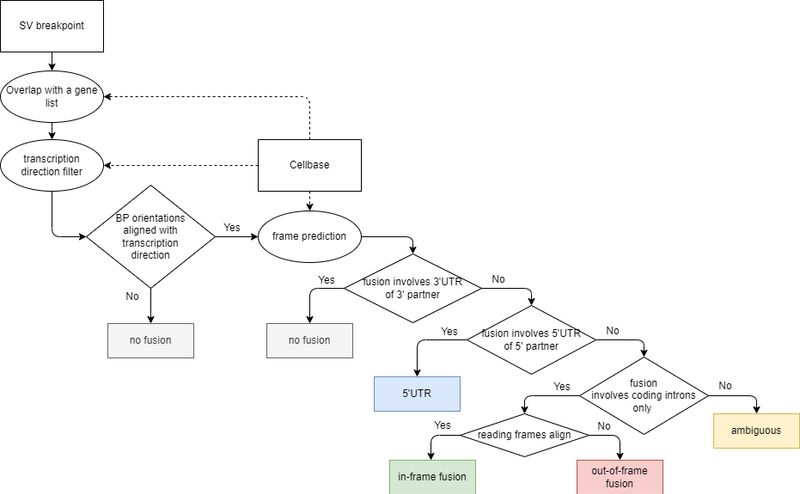
Figure 4. Fusion outcome decision scheme
Ambiguous outcome is the biggest class of fusions; it covers all the cases where it is not possible to predict the frame.
For example, if a fusion involves 5’ UTR of the downstream partner (the region just upstream of the coding sequence), it is hard to predict how the chimeric pre-mRNA will be spliced. The resulting fusion transcript may contain a part of the downstream partner’s first exon or may omit it completely (more information about splicing mechanisms can be read here).
Similarly, if a fusion affects 3’ UTR of the upstream partner (e.g., in the case of BCOR-CCNB3 fusions in sarcomas), it may be unproductive if the terminal codon remains readable on the upstream side after splicing, or productive if not. If a variant affects a coding exon or a splice site, it is very hard to confidently predict what is going to happen.
What’s next?
Hooray! We now have the potential outcome annotations.
For each structural variant in the pipeline’s output, a list of potential fusion pairs with their outcomes will be reported (Figure 5). Now the number of fusions to explore further is much smaller. For example, in several sarcoma samples that we tested, only 5% of all potential DNA fusions were annotated as potentially productive (those annotated as 5’ UTR, in-frame, or ambiguous).
Providing predictions of the potential fusion outcome spares the need to research transcription orientations and exon phases of the fusion partners when interpreting the WGA results.
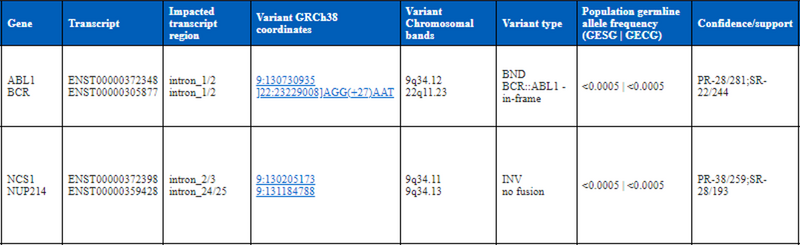
Figure 5. Appearance of potential fusion outcome annotation in the whole genome analysis reports
Moreover, our approach relies on the precision of breakpoint locations, which likewise, is not always perfect.
As the annotation is performed separately for each breakpoint, there isn’t currently a means of resolving the fusion structure for complex variants that involve multiple breakpoints. Some genes can be read into multiple different transcripts, and it would require much more computational resources to assess and present productivity for all possible combinations.
As such, we currently restrict ourselves to just the canonical ones.
It is also worth admitting that we simplified the task by throwing as many corner cases as possible under the ‘ambiguous’ outcome hood.
In fact, there are currently no methods available to confidently predict fusion outcome from WGS data when breakpoints hit exons or splice sites. There are known cases when a translocation between exons of different genes can activate cryptic splice sites and completely alter the resulting transcript. Though even in the ‘simple’ cases when 2 introns between exons are fused and the fusion is seemingly out-of-frame, it is possible that one of the surrounding exons will be omitted. This would cause an in-frame fusion to be generated and is known as “exon skipping”.
We plan to implement an additional check for this in the future. Hopefully, future researchers will be able to utilise increasing computational resources and more sophisticated algorithms to completely solve DNA-based fusion prediction.
There is more to a cancer variant that produces a fusion than whether it is in-frame or out-of-frame. We need to know whether a variant is present in a large portion of the tumour or not, and it matters which exons are included in the fusion from either side.
All this information is also present in the whole genome analysis results, allowing users to further prioritise the findings. Depending on the precise fusion and patient’s tumour type, its discovery via whole genome analysis may lead to changes in diagnosis or patient care, ultimately resulting in better outcomes for patients.

