Scouring the scientific literature with large language models
Curating evidence of gene-disease associations
By Sam Barnett, Arina Puzriakova, Catherine Snow, Eleanor Williams, and Francisco Azuaje onWe have developed a machine learning tool for scanning large volumes of scientific literature to find reports of gene-disease associations using AWS Cloud services and Large Language Models. This will help Genomics England to use the most up-to-date knowledge of rare disease genomics in our diagnostic pipelines, ensuring that patients are getting the most accurate results.
Collecting evidence of gene-disease associations
When using whole genome sequencing (WGS) to investigate the genomic cause of a patient’s condition, we aim to provide the clinical scientists with a manageable list of potentially causative variants to be prioritised for further analysis. Typically, WGS identifies around 6,500,000 variants per patient that are different to the reference genome. Thus, candidate variants must be selected using a number of criteria, including whether the variant is in a gene that is already known to be associated with a disease that has a similar phenotypic profile to that of the patient.
To facilitate this prioritisation process, Genomics England created a knowledgebase called PanelApp. This links genes to diseases by grouping them into panels relating to clinical indications. Genes on a panel are classified as red, amber or green; depending on the amount of evidence supporting the association with a particular disease. Only green genes have a diagnostic level of evidence.
The aim is to search for new evidence that could result in a “not-yet-reportable" red and amber gene being promoted to green, allowing it to be used in patient analysis. More details on the gene curation and panel update process can be found in a previous blog.

Figure 1: A depiction of a Panel (left) demonstrating genes with different levels of evidence. Different sources of evidence for assessing a gene-disease association (right) with new publications highlighted as the focus of this work.
The Biocuration team are a vital part of the evidence discovery process. They gather information from many different sources including feedback from clinicians, Genomics England’s diagnostic discovery team, scientific publications and other databases.
While scientific articles provide a vital source of primary data, each year more than 3 million scientific articles are published. Manual monitoring of this vast ocean of documents is therefore unfeasible. The Biocuration team currently mainly rely on literature searches and publications being highlighted to the team by different stakeholders. However, it would be beneficial to have an efficient method for identifying publications relevant to red and amber genes.
The machine learning workflow
We approached this problem by starting with a static snapshot of scientific literature as published in the Public Library of Science (PLoS – API access shown here). This was a relatively manageable corpus of almost 350,000 articles in which we know there are positive examples present.
We first cleaned up the text by removing all the metadata and references, as well as all the JATS XML tags to leave just the body of the scientific text.
The second stage was to find mentions of genes in papers and match specific literature to a specific gene to make a sub-corpus for each gene. This could be achieved with a simple keyword search approach due to the commonly adopted HGNC standardised gene names.
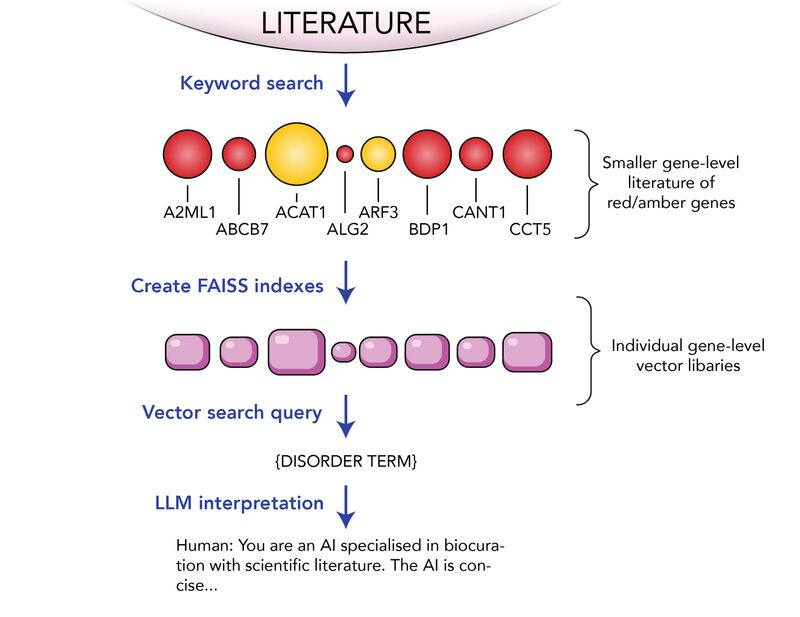
Figure 2: Strategy for finding gene-disease associations in the scientific literature.
The third stage was to generate numerical embeddings for all the articles associated with each gene. Each article was chunked into smaller pieces of text and passed through a machine learning model (Amazon’s Titan) to capture the semantic meaning the text.
The embeddings are then stored in a vector library, in our case powered by FAISS developed by Meta. At this point we have many vector libraries, one for each gene. We can query each one with an embedding (also generated with Titan) representing a panel name such as ‘Intellectual disability’, using cosine similarity to find articles that are similar in content to our search term. We chose to create separate vector libraries for each gene so that we could also reuse them in cases where the same genes occur in different panels.
We now have a small set of papers that mention a gene and also have a semantic similarity with a panel name of interest. The final step is to submit the paper to a large Language Model, in this case Anthropic’s Claude 2.1, to determine if there is any primary evidence for a gene-disease association in the article. If Claude determines that there is evidence, we ask it to generate a summary of the evidence in the paper. This evidence can be used by our Biocuration experts to help decide if it is a paper worth reading in detail.
Prompt engineering
To achieve the best results from Claude we went through several rounds of prompt engineering, tweaking the prompt in response to results. Our current prompt can be seen in the figure below with a breakdown of why each section in the prompt was present.
For example, we wanted Claude to generate an output even if it thinks that the evidence is inconclusive, as the ultimate arbiter for the evidence should be the Biocuration team and Claude was sometimes overly cautious. We also realised that Claude appeared not to be aware of all of a gene’s aliases, so we explicitly include them in the prompt.
We also wanted Claude to include information as to whether the information is from a reference to another source, as these articles are less of a priority than primary evidence.

Figure 3: Prompt that was used for this work with explanations for why sections were included.
Results
For speed purposes, we initially focused on the first run on red genes on the Intellectual Disability panel, as well as genes with fewer than 100 associated papers in the PLoS corpus. We surfaced 20 candidate papers which contained information relating to both our genes and the phenotype of interest. Rationale for prioritising each paper was compressed into 1-4 sentence summaries, providing a powerful insight for the Biocurator who could preliminarily assess the extent of available evidence before delving into the full text.
Curation of the 20 candidate papers revealed 2 genes that had sufficient evidence to be promoted from red to green, meaning they could be used in future patient analysis. Additional unrelated cases, reported in other publications, were used to corroborate the association and fulfil the criteria for a gene rating upgrade to green status. However, the machine learning pipeline was useful for finding papers that reinforced pre-existing evidence in PanelApp, and prompted further investigation for additional studies to ultimately allow for gene promotion.
2 genes were promoted from red to amber, meaning that we were able to identify additional support for their involvement in intellectual disability disorders, but the evidence is insufficient to conclusively make the link.
Red gene ratings were maintained following curation of the remaining 16 publications, but this still provided valuable information:
- We identified a paper that was previously missed by manual methods due to results being buried within the article. These would not have been easily recognised as relevant based on the title or abstract alone.
- 2 papers had inconclusive findings due to patients having variants in other genes that could also plausibly explain their condition. Nonetheless, these reports could become important for future evaluations if more similarly affected patients come to light.
- Seven publications contained relevant evidence but had already been reviewed in PanelApp, without any new discoveries made since. This shows that the machine learning tool we have developed is able to detect relevant publications, which the Biocuration team can use to assess genetic causes of conditions
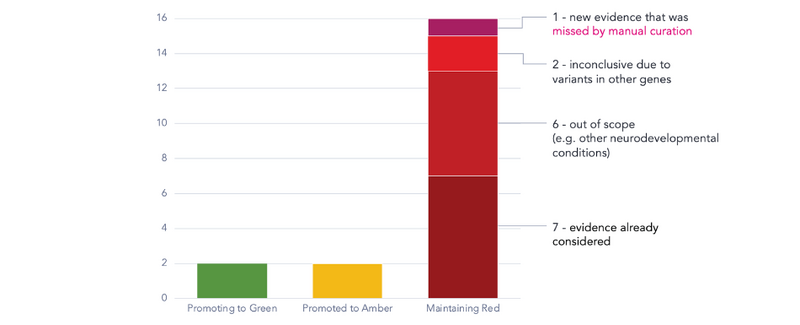
Figure 4: Outcome of curation of 20 candidate papers identified by the machine learning workflow applied to red genes on the Intellectual disability panel.
Compared to manual searches of bibliographic databases, such as PubMed and biomedical journals, our machine learning approach proved superior for rapid scanning of large volumes of scientific literature to extract multiple papers for assessing gene-disease relationships. This project demonstrates that emerging Generative AI techniques can be used to complement Biocuration to ease the workload and increase efficiency.
Next steps
We are in the process of productionising this solution, meaning we are taking it to the level of live service, to actively scan newly published scientific literature. This can then be regularly delivered to the Biocuration team.
This will allow Genomics England to keep pace with the latest advancements in scientific understanding and update gene panels for the Genomic Medicine Service. This will ultimately lead to faster, more accurate diagnoses and improve outcomes for patients.
Acknowledgements
This work was done in tandem with the AWS Healthcare data science team who provided valuable input into the design of the workflow. They contributed key ideas to the design so that we achieved the best possible results. We also thank AWS for granting us preview access to the Bedrock service for this work, as well as our Genomics England colleagues for supporting this work and assisting with the writing and editing of this blog.

